In 2020 the Nobel prize for medicine was awarded to Harvey Alter, Michael Houghton and Charles Rice for their role in the discovery of the hepatitis C virus. I now watched the videos of the corresponding Nobel lectures. For my taste the lecture of Alter was by far the most interesting of the three. I think that he was also the one who played the most fundamental role in this discovery. At the beginning of his lecture he emphasizes the point that the most important discoveries in science often come as a complete surprise and not as a result of planned research programmes. Alter was 85 when he got the prize and so he had to wait a long time for it. The papers documenting his fundamental contributions were published in 1989. A central part of this work was the collection and preservation of blood samples from patients undergoing open heart surgery. Why was this group chosen? One of the most important modes of infection with hepatitis B and C used to be blood transfusions. This continued to be the case until tests were available to screen donors for these diseases. This kind of surgery involves extensive blood transfusions and so the chances of infection were relatively high in these patients. Also these patients suffered from relatively few other diseases which could have been confounding factors. These blood samples were an invaluable resource in the search for the virus. They were the basis of painstaking analysis over many years.
One important feature of hepatitis C is that it becomes chronic in 70 per cent of cases. This looks like a failure of the immune system to handle this disease. What are the reasons for this failure? One concerns quasispecies. The hepatitis C virus has an RNA genome and the copying of RNA is very error-prone. This leads to a huge variety in the genomes of virions in a single patient. This in turn results in rapid mutations of the virus. If an antibody has developed to combat the virus then selective pressure will quickly cause a new form to become dominant which is not vulnerable to that antibody. It seems to me that if this type of effect is to be captured using mathematical model it will require a stochastic model. Deterministic models of the type I have studied in the past are probably not helpful for that. In the lecture it is also mentioned that the number of T cells (CD4+ and CD8+) declines very much in chronically infected hepatitis C patients. No explanation is offerred as to why that is the case. Deterministic mathematical models might be able to contribute some understanding in that case.
The lecture contains the following interesting story. There was a time at which liver cancer was much more common in Japan than in the West. The reason for this was that that cancer was in many cases a late stage effect of hepatitis C. During wars in the early part of the 20th century many Japanese soldiers injected drugs with shared needles and this was what spread the disease. It was observed that there were many cases of jaundice (the most striking symptom of hepatitis) on the battlefield. Decades later many of these men developed serious liver disease, including cancer. Japanese doctors predicted that a similar phenomenon would be seen in the West when the effects of recreational drug use became manifest. They were right.
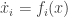 . It should be defined on the
. It should be defined on the 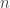 -dimensional Euclidean space or on one of its orthants. (An orthant is the subset of Euclidean space defined by making a choice of the signs of its components. It generalises a quadrant in the two-dimensional case.) The system is said to be cooperative if
-dimensional Euclidean space or on one of its orthants. (An orthant is the subset of Euclidean space defined by making a choice of the signs of its components. It generalises a quadrant in the two-dimensional case.) The system is said to be cooperative if  for all
for all  . The name comes from the fact that the equations for the population dynamics of a set of species has this property if each species benefits the others. Suppose we now have two solutions
. The name comes from the fact that the equations for the population dynamics of a set of species has this property if each species benefits the others. Suppose we now have two solutions  and
and 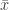 of the system and that
of the system and that  for all
for all 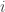 at some time time
at some time time 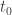 . We may abbreviate this relation by
. We may abbreviate this relation by 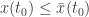 . Here we see a partial order on Euclidean space defined by the ordering of the components. A theorem of Müller and Kamke says that if the initial data for two solutions of a cooperative system at time
. Here we see a partial order on Euclidean space defined by the ordering of the components. A theorem of Müller and Kamke says that if the initial data for two solutions of a cooperative system at time 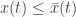 for all
for all 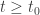 . Another way of saying this is that the time-
. Another way of saying this is that the time-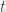 flow of the system is preserves the partial order. A system of ODE with this property is called monotone. Thus the Müller-Kamke theorem says that a cooperative system is monotone.
flow of the system is preserves the partial order. A system of ODE with this property is called monotone. Thus the Müller-Kamke theorem says that a cooperative system is monotone.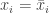 for a certain
for a certain 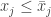 for
for  then
then  . To see this we join
. To see this we join 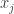 to
to  . On each segment of this curve the value of
. On each segment of this curve the value of 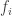 does not decrease, as a consequence of the fundamental theorem of calculus. Hence its value at the end of the entire path is at least as big as its value at the beginning. We now want to prove that a certain inequality holds at all times
does not decrease, as a consequence of the fundamental theorem of calculus. Hence its value at the end of the entire path is at least as big as its value at the beginning. We now want to prove that a certain inequality holds at all times 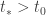 where the inequality fails and get a contradiction. Unfortunately there might be no such time – in principle the condition might fail immediately. To get around this we deform the system for the solution
where the inequality fails and get a contradiction. Unfortunately there might be no such time – in principle the condition might fail immediately. To get around this we deform the system for the solution 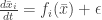 . If we can prove the result for the deformed system the result for the initial system follows by continuous dependence of the solution on
. If we can prove the result for the deformed system the result for the initial system follows by continuous dependence of the solution on  . For the deformed system let
. For the deformed system let 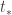 be the supremum of the times where the desired inequality holds. If the inequality does not hold globally then the system is still defined at
be the supremum of the times where the desired inequality holds. If the inequality does not hold globally then the system is still defined at  . For
. For 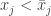 for some
for some 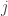 since otherwise the two solutions would be equal and the result trivial. The integrated form of the cooperativity condition implies that at
since otherwise the two solutions would be equal and the result trivial. The integrated form of the cooperativity condition implies that at 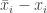 is positive. On the other hand the fact that it just reached zero coming from positive values implies that the right hand side of the evolution equation is non-positive and we get a contradiction.
is positive. On the other hand the fact that it just reached zero coming from positive values implies that the right hand side of the evolution equation is non-positive and we get a contradiction.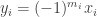 where each of the
where each of the 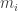 are zero or one. This transforms the signs of
are zero or one. This transforms the signs of 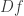 in a certain way and so cooperativity of the system for
in a certain way and so cooperativity of the system for 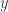 corresponds to a certain sign pattern for the entries of
corresponds to a certain sign pattern for the entries of  should be either non-negative or non-positive. Next, the sign of
should be either non-negative or non-positive. Next, the sign of  is not changed be the transformation and must thus be non-negative. In the context of population models this can be interpreted as saying that there is no pair of species which are in a predator-prey relationship. Given that these two conditions are satisfied we consider a labelled graph where the nodes are the numbers from
is not changed be the transformation and must thus be non-negative. In the context of population models this can be interpreted as saying that there is no pair of species which are in a predator-prey relationship. Given that these two conditions are satisfied we consider a labelled graph where the nodes are the numbers from 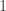 to
to  . The transformation
. The transformation  makes it into a competitive system. In various models of virus dynamics including the immune response the target cells of the virus and the immune cells are in a predator-prey relationship and so these systems can be neither cooperative up to sign or competitive up to sign.
makes it into a competitive system. In various models of virus dynamics including the immune response the target cells of the virus and the immune cells are in a predator-prey relationship and so these systems can be neither cooperative up to sign or competitive up to sign.