Here I consider a system of reaction-diffusion equations of the form  ,
,  . The functions
. The functions  are defined on
are defined on 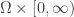 where
where 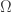 is a bounded domain in
is a bounded domain in 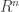 with smooth boundary. Let the
with smooth boundary. Let the 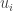 be denoted collectively by
be denoted collectively by  . I assume that the diffusion coefficients
. I assume that the diffusion coefficients 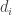 are non-negative. If some of them are zero then the system is degenerate. In particular there is an ODE special case where all are zero. If this system really describes chemical reactions and the are concentrations then it is natural to assume that
are non-negative. If some of them are zero then the system is degenerate. In particular there is an ODE special case where all are zero. If this system really describes chemical reactions and the are concentrations then it is natural to assume that  for all
for all 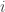 . It should then follow that in the presence of suitable boundary conditions
. It should then follow that in the presence of suitable boundary conditions  for all
for all 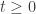 . I assume that is a classical solution and that it extends to the boundary with enough smoothness that the boundary conditions are defined pointwise. It is necessary to implement the idea that the system is defined by chemical reactions. This can be done by requiring that whenever
. I assume that is a classical solution and that it extends to the boundary with enough smoothness that the boundary conditions are defined pointwise. It is necessary to implement the idea that the system is defined by chemical reactions. This can be done by requiring that whenever  and
and 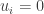 it follows that
it follows that 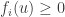 . (This means that if a chemical species is not present it cannot be consumed in any reaction.) It turns out that this condition is enough to ensure positivity.
. (This means that if a chemical species is not present it cannot be consumed in any reaction.) It turns out that this condition is enough to ensure positivity.
I will now explain a proof of positivity. The central ideas are taken from a paper of Maya Mincheva and David Siegel (J. Math. Chem. 42, 1135). Thanks to Maya for helpful comments on this subject. The argument is already interesting for ODE since important conceptual elements can already be seen. I will first discuss that case. Consider a solution of the equation  on the interval
on the interval 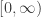 where
where  and
and 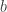 are continuous functions with
are continuous functions with  and suppose that
and suppose that  . I claim that
. I claim that  for all . Let
for all . Let ![t^*=\sup\{t_1:u(t)\ge 0\ {\rm on}\ [0,t_1]\}](https://s0.wp.com/latex.php?latex=t%5E%2A%3D%5Csup%5C%7Bt_1%3Au%28t%29%5Cge+0%5C+%7B%5Crm+on%7D%5C+%5B0%2Ct_1%5D%5C%7D&bg=ffffff&fg=333333&s=0&c=20201002) . Since it follows by continuity that
. Since it follows by continuity that  . Assume that
. Assume that  . By continuity
. By continuity  . If
. If 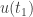 were greater than zero then by continuity it would also be positive for
were greater than zero then by continuity it would also be positive for 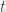 slightly larger than
slightly larger than 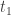 , contradicting the definition of . Thus
, contradicting the definition of . Thus  . The evolution equation then implies that
. The evolution equation then implies that  . This implies that
. This implies that  for slightly less than , which also contradicts the definition of . Hence in reality
for slightly less than , which also contradicts the definition of . Hence in reality 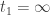 and this completes the proof of the desired result.
and this completes the proof of the desired result.
Suppose now that we weaken the assumptions to  and
and  . We would like to conclude that for all . To do this we define a new quantity
. We would like to conclude that for all . To do this we define a new quantity  for positive constants
for positive constants  and
and 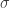 . Then
. Then 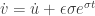 . Hence
. Hence ![\dot v=-au+[b+\epsilon\sigma e^{\sigma t}]](https://s0.wp.com/latex.php?latex=%5Cdot+v%3D-au%2B%5Bb%2B%5Cepsilon%5Csigma+e%5E%7B%5Csigma+t%7D%5D&bg=ffffff&fg=333333&s=0&c=20201002) and
and  . Now
. Now  and so
and so ![\dot v=-av+[b+\epsilon(\sigma+a) e^{\sigma t}]](https://s0.wp.com/latex.php?latex=%5Cdot+v%3D-av%2B%5Bb%2B%5Cepsilon%28%5Csigma%2Ba%29+e%5E%7B%5Csigma+t%7D%5D&bg=ffffff&fg=333333&s=0&c=20201002) . It follows that if is large enough
. It follows that if is large enough  satisfies the conditions satisfied by in the previous argument and it can be concluded that
satisfies the conditions satisfied by in the previous argument and it can be concluded that  for all . Letting tend to zero shows that for all , the desired result.
for all . Letting tend to zero shows that for all , the desired result.
This is different, and perhaps a bit more complicated than, the proof I know for this type of result. That proof involves considering the derivative of  on
on 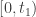 . It also involves approximating non-negative data by positive data. A difference is that the proof just given does not use the continuous dependence of solutions of an ODE on initial data and in that sense it is more elementary. In Theorem 3 of the paper of Mincheva and Siegel the former proof is extended to a case involving a system of PDE.
. It also involves approximating non-negative data by positive data. A difference is that the proof just given does not use the continuous dependence of solutions of an ODE on initial data and in that sense it is more elementary. In Theorem 3 of the paper of Mincheva and Siegel the former proof is extended to a case involving a system of PDE.
Now I come to that PDE proof. The system of PDE concerned is the one introduced above. Actually the paper requires the be positive but that stronger condition is not necessary. This equation is solved with an initial datum  and a boundary condition
and a boundary condition 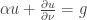 . Here
. Here 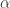 is a diagonal matrix with non-negative entries and the function
is a diagonal matrix with non-negative entries and the function  is non-negative. The derivative
is non-negative. The derivative 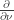 is that in the direction of the outward unit normal to the boundary. We assume that is a classical solution, i.e. all derivatives of appearing in the equation exist and are continuous. Moreover has a continuous extension to
is that in the direction of the outward unit normal to the boundary. We assume that is a classical solution, i.e. all derivatives of appearing in the equation exist and are continuous. Moreover has a continuous extension to 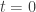 and a
and a 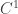 extension to
extension to  . We now replace the differential equation by the differential inequality
. We now replace the differential equation by the differential inequality  . We assume that the initial data are non-negative,
. We assume that the initial data are non-negative, 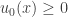 . The assumption that is non-negative, together with the boundary condition, gives rise to the inequality
. The assumption that is non-negative, together with the boundary condition, gives rise to the inequality  . The aim is to show that all solutions of the resulting system of inequalities are non-negative. We assume the condition for a system of chemical reactions already mentioned.
. The aim is to show that all solutions of the resulting system of inequalities are non-negative. We assume the condition for a system of chemical reactions already mentioned.
The proof is a generalization of that already given in the ODE case. The first step is to treat the case where each of the inequalities is replaced by the corresponding strict inequality. In contrast to the proof in the paper we assume that that 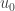 is strictly positive on
is strictly positive on  . We define as in the ODE case so that
. We define as in the ODE case so that ![[0,t_1]](https://s0.wp.com/latex.php?latex=%5B0%2Ct_1%5D&bg=ffffff&fg=333333&s=0&c=20201002) is the longest interval where the solution is non-negative. We suppose that is finite and obtain a contradiction. Note first that, as in the ODE case, by continuity. Now . If were strictly positive on then by continuity would be strictly positive for slightly greater than , contradicting the definition of . Hence there is an index and a point
is the longest interval where the solution is non-negative. We suppose that is finite and obtain a contradiction. Note first that, as in the ODE case, by continuity. Now . If were strictly positive on then by continuity would be strictly positive for slightly greater than , contradicting the definition of . Hence there is an index and a point  with
with 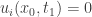 and
and 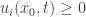 for all
for all 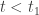 . Suppose first that
. Suppose first that  . Then
. Then  and
and  . This contradicts the strict inequality related to the evolution equation for and so cannot happen. Suppose next that
. This contradicts the strict inequality related to the evolution equation for and so cannot happen. Suppose next that 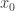 is on the boundary of . Then
is on the boundary of . Then 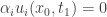 and
and  . This contradicts the strict inequality related to the boundary condition for . Thus in fact and is strictly positive for all time.
. This contradicts the strict inequality related to the boundary condition for . Thus in fact and is strictly positive for all time.
Now we do a perturbation argument by considering 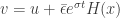 . Here
. Here 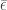 is the vector all of whose components are and
is the vector all of whose components are and  is a positive function. It is obvious that
is a positive function. It is obvious that 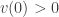 . We now choose
. We now choose  which ensures its positivity and require that the outward normal derivative of
which ensures its positivity and require that the outward normal derivative of 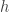 on the boundary is equal to one. (Here I will take for granted that a function of this kind exists. A source for this statement is cited in the paper. For me the positivity statement is already very interesting in the case that is a ball and there the existence of the function is obvious.) Then the fact that satisfies the non-strict boundary inequality implies that satisfies the strict boundary inequality. It remains to derive an evolution equation for . A straightfoward calculation gives
on the boundary is equal to one. (Here I will take for granted that a function of this kind exists. A source for this statement is cited in the paper. For me the positivity statement is already very interesting in the case that is a ball and there the existence of the function is obvious.) Then the fact that satisfies the non-strict boundary inequality implies that satisfies the strict boundary inequality. It remains to derive an evolution equation for . A straightfoward calculation gives ![\frac{\partial v_i}{\partial t}-d_i\Delta v_i-f_i(v)\ge e^{\sigma t}\epsilon[\sigma H-d_i\Delta H-L\sqrt{n}H]](https://s0.wp.com/latex.php?latex=%5Cfrac%7B%5Cpartial+v_i%7D%7B%5Cpartial+t%7D-d_i%5CDelta+v_i-f_i%28v%29%5Cge+e%5E%7B%5Csigma+t%7D%5Cepsilon%5B%5Csigma+H-d_i%5CDelta+H-L%5Csqrt%7Bn%7DH%5D&bg=ffffff&fg=333333&s=0&c=20201002) where
where 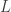 is a Lipschitz constant for
is a Lipschitz constant for 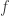 on the image of the compact region being considered. Choosing large enough ensures that the right hand side and hence the left hand side of this inequality is strictly positive. We conclude that
on the image of the compact region being considered. Choosing large enough ensures that the right hand side and hence the left hand side of this inequality is strictly positive. We conclude that  and, letting
and, letting  , that .
, that .
If we want to prove an inequality for solutions of a PDE it is common to proceed as follows. We deform the problem continuously as a function of a small parameter so as to get a simpler problem. When that has been solved we let tend to zero to get a solution of the original problem. Often it is the equations which are deformed. Then we need a theorem on existence and continuous dependence to get a continuous deformation of the solution. The above proof is different. We perturb the solution in a way whose continuity is obvious and then derive a family of equations of which that is a family of solutions. This is easy and comfortable. The hard thing is to guess a good deformation of the soluion.

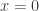

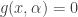





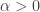

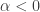








 , where
, where  is the number of susceptibles and
is the number of susceptibles and 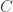 is the concentration of virus in the environment which may cause infection. What should we choose for the function
is the concentration of virus in the environment which may cause infection. What should we choose for the function  and the Hill function where
and the Hill function where  . The Michaelis-Menten function has a mechanistic basis, introduced by the people who it is now named after. Hill also presented a mechanistic basis for his function. It incorporates the phenomenon of cooperative binding, with the original example being the binding of oxygen to haemoglobin. Another example of the choice of response functions occurs in predator-prey models in ecology. Here
. The Michaelis-Menten function has a mechanistic basis, introduced by the people who it is now named after. Hill also presented a mechanistic basis for his function. It incorporates the phenomenon of cooperative binding, with the original example being the binding of oxygen to haemoglobin. Another example of the choice of response functions occurs in predator-prey models in ecology. Here 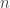 as in Holling type II (
as in Holling type II (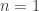 ) and Holling type III (
) and Holling type III ( ). We found that the case
). We found that the case 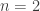 gives rise to backward bifurcations while the case
gives rise to backward bifurcations while the case  ,
, 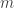 of these have the status of infected variables. The right hand side of the evolution equation for
of these have the status of infected variables. The right hand side of the evolution equation for 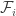 ,
,  and
and  where
where  are all non-negative. This is condition (A1) of the paper. On the right hand side of this type of system there are positive and negative terms. If we take
are all non-negative. This is condition (A1) of the paper. On the right hand side of this type of system there are positive and negative terms. If we take  is also used. As is usual in reaction networks
is also used. As is usual in reaction networks  when
when  . This is condition (A2) of the paper. The
. This is condition (A2) of the paper. The  . This is condition (A3) of the paper. It restricts the ambiguity in the splitting. In condition (A4) the set of disease-free steady states is supposed to be invariant and this leads to the vanishing of some non-negative terms at those points. In general this condition restricts the choice of the infected compartments. In the fundamental model of virus dynamics the infected variables are a subset of
. This is condition (A3) of the paper. It restricts the ambiguity in the splitting. In condition (A4) the set of disease-free steady states is supposed to be invariant and this leads to the vanishing of some non-negative terms at those points. In general this condition restricts the choice of the infected compartments. In the fundamental model of virus dynamics the infected variables are a subset of 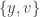 . However choosing it to be a proper subset would violate condition (A4). Thus for this model there is no choice at this point. There does remain a choice in splitting the positive terms, as already discussed in a
. However choosing it to be a proper subset would violate condition (A4). Thus for this model there is no choice at this point. There does remain a choice in splitting the positive terms, as already discussed in a 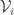 also splits the linearization into a sum. It is an elementary consequence of the assumptions made that some of the entries of the matrices occurring in these decompositions are zero. It can also be shown that some entries are non-negative. To describe a new infection we can take an initial datum close to the disease-free steady state. Then we may reasonably suppose (leaving aside questions of proof) that the actual dynamics is well-approximated by the linearized dynamics during an initial period. Under the given assumptions the linearization is block lower triangular. The linearized evolution is partially decoupled and the uninfected variables decay exponentially. So the dynamics essentially reduces to that of the infected variables and is generated by the upper left block, called
also splits the linearization into a sum. It is an elementary consequence of the assumptions made that some of the entries of the matrices occurring in these decompositions are zero. It can also be shown that some entries are non-negative. To describe a new infection we can take an initial datum close to the disease-free steady state. Then we may reasonably suppose (leaving aside questions of proof) that the actual dynamics is well-approximated by the linearized dynamics during an initial period. Under the given assumptions the linearization is block lower triangular. The linearized evolution is partially decoupled and the uninfected variables decay exponentially. So the dynamics essentially reduces to that of the infected variables and is generated by the upper left block, called  in the paper. We get an exponential phase of decay with populations proportional to
in the paper. We get an exponential phase of decay with populations proportional to 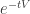 . During this phase the number of susceptibles is approximately constant, equal to its value at the disease-free steady state. Hence we get an explicit expression for the rate of infection in this phase. Integrating this with respect to time gives an expression for the ratio of total number of individuals infected in that phase as a function of the original number of individuals in the different compartments. It is given by the matrix
. During this phase the number of susceptibles is approximately constant, equal to its value at the disease-free steady state. Hence we get an explicit expression for the rate of infection in this phase. Integrating this with respect to time gives an expression for the ratio of total number of individuals infected in that phase as a function of the original number of individuals in the different compartments. It is given by the matrix  , which is the NGM previously mentioned.
, which is the NGM previously mentioned.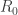 is simply interpreted as the known combination of parameters. After I had written this I noticed a source cited by van den Driessche and Watmough, the book ‘Mathematical Epidemiology of Infectious Diseases’ by Diekmann and Heesterbeek and, in particular, Theorem 6.13 of that book. (To avoid any confusion, I am talking about the original edition of this book, not the later extended version.) I had never seen the book before and I borrowed it from the library. I found a text which pays attention to the relations between mathematical rigour and applications in an exemplary way. I feel that if I read the whole book carefully and did all the exercises (which is the recommendation of the authors) I might be able to get rid of all the problems which are the subject of this and the previous related posts. Since this ideal engagement with the book is something I will not achieve soon, if ever, I restrict myself to some limited comments. On p. 105 the authors write ‘Many modellers … even distrust whether the threshold value one of
is simply interpreted as the known combination of parameters. After I had written this I noticed a source cited by van den Driessche and Watmough, the book ‘Mathematical Epidemiology of Infectious Diseases’ by Diekmann and Heesterbeek and, in particular, Theorem 6.13 of that book. (To avoid any confusion, I am talking about the original edition of this book, not the later extended version.) I had never seen the book before and I borrowed it from the library. I found a text which pays attention to the relations between mathematical rigour and applications in an exemplary way. I feel that if I read the whole book carefully and did all the exercises (which is the recommendation of the authors) I might be able to get rid of all the problems which are the subject of this and the previous related posts. Since this ideal engagement with the book is something I will not achieve soon, if ever, I restrict myself to some limited comments. On p. 105 the authors write ‘Many modellers … even distrust whether the threshold value one of  . Beyond this, I believe that
. Beyond this, I believe that  , a fact with obvious consequences for stability of that steady state. It is clearly stated in the paper that the NGM does not only depend on the system of ODE defining the model but also on the choices already mentioned. The first choice is that of the
, a fact with obvious consequences for stability of that steady state. It is clearly stated in the paper that the NGM does not only depend on the system of ODE defining the model but also on the choices already mentioned. The first choice is that of the 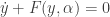 depending on a parameter
depending on a parameter 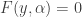 . Sometimes we can eliminate all but one of the variables to obtain an equation
. Sometimes we can eliminate all but one of the variables to obtain an equation  whose solutions are in one to one correspondence with those of the original equation for steady states. Clearly this situation is closely related to Lyapunov-Schmidt reduction in the case where the linearization has corank one. Often the reduced equation is much easier to treat that the original one and this can be used to obtain information on the number of steady states of the ODE system. This can be used to study multistationarity in systems of ODE arising as models in molecular biology. In that context we would like more refined information related to multistability. In other words, we would like to know something about the stability of the steady states produced by the reduction process. Stablity is a dynamical property and so it is not a priori clear that it can be investigated by looking at the equation for steady states on its own. Different ODE systems can have the same set of steady states. Note, however, that in the case of hyperbolic steady states the stability of a steady state is determined by the eigenvalues of the linearization of the function
whose solutions are in one to one correspondence with those of the original equation for steady states. Clearly this situation is closely related to Lyapunov-Schmidt reduction in the case where the linearization has corank one. Often the reduced equation is much easier to treat that the original one and this can be used to obtain information on the number of steady states of the ODE system. This can be used to study multistationarity in systems of ODE arising as models in molecular biology. In that context we would like more refined information related to multistability. In other words, we would like to know something about the stability of the steady states produced by the reduction process. Stablity is a dynamical property and so it is not a priori clear that it can be investigated by looking at the equation for steady states on its own. Different ODE systems can have the same set of steady states. Note, however, that in the case of hyperbolic steady states the stability of a steady state is determined by the eigenvalues of the linearization of the function  at that point. Golubitsky and Schaeffer prove the following remarkable result. (It seems clear that results of this kind were previously known in some form but I did not yet find an earlier source with a clear statement of this result free from many auxiliary complications.) Suppose that we have a bifurcation point
at that point. Golubitsky and Schaeffer prove the following remarkable result. (It seems clear that results of this kind were previously known in some form but I did not yet find an earlier source with a clear statement of this result free from many auxiliary complications.) Suppose that we have a bifurcation point  where the linearization of
where the linearization of 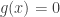 and
and  then for
then for  . Thus the stability of steady states arising at the bifurcation point is determined by the function
. Thus the stability of steady states arising at the bifurcation point is determined by the function 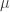 , is a smooth function of
, is a smooth function of  is also a smooth function. The aim is to show that they have the same sign. This would be enough to prove the desired stability statement. Suppose that the gradient of
is also a smooth function. The aim is to show that they have the same sign. This would be enough to prove the desired stability statement. Suppose that the gradient of  and letting
and letting  . Lyapunov-Schmidt reduction is applied to
. Lyapunov-Schmidt reduction is applied to  to get a function
to get a function  . Let
. Let  be the eigenvalue of
be the eigenvalue of 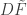 which is analogous to the eigenvalue
which is analogous to the eigenvalue  . From what was said above it follows that, in a notation which is hopefully clear,
. From what was said above it follows that, in a notation which is hopefully clear,  implies
implies  . We now want to show that the gradients of these two functions are non-zero. Lyapunov-Schmidt theory includes a formula expressing
. We now want to show that the gradients of these two functions are non-zero. Lyapunov-Schmidt theory includes a formula expressing  in terms of
in terms of  . Next we turn to the gradient of
. Next we turn to the gradient of  , more specifically to the derivative of
, more specifically to the derivative of 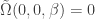 for all
for all 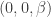 shows that
shows that 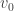 is an eigenvalue of
is an eigenvalue of  for all
for all 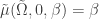 and the derivative of
and the derivative of  with respect to
with respect to 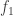 and
and 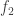 are two smooth functions,
are two smooth functions,  extends smoothly to the zero set of
extends smoothly to the zero set of  with
with ![\tilde a(0,0,0)=[\frac{\partial}{\partial\beta}(\tilde\Omega,0,0)]/\tilde g_{x\beta}(0,0,0)]>0](https://s0.wp.com/latex.php?latex=%5Ctilde+a%280%2C0%2C0%29%3D%5B%5Cfrac%7B%5Cpartial%7D%7B%5Cpartial%5Cbeta%7D%28%5Ctilde%5COmega%2C0%2C0%29%5D%2F%5Ctilde+g_%7Bx%5Cbeta%7D%280%2C0%2C0%29%5D%3E0&bg=ffffff&fg=333333&s=0&c=20201002) . Setting
. Setting 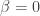 in the first equation then gives the desired conclusion.
in the first equation then gives the desired conclusion. . It was left open, whether this is true for all choices of parameters. In the second paper we show that it is not: there are parameters for which periodic solutions exist. This is proved by demonstrating the presence of Hopf bifurcations. These are obtained by a perturbation argument starting from a simpler model. Unfortunately we could not decide analytically whether the periodic solutions are stable or unstable. Simulations indicate that they are stable at least in some cases.
. It was left open, whether this is true for all choices of parameters. In the second paper we show that it is not: there are parameters for which periodic solutions exist. This is proved by demonstrating the presence of Hopf bifurcations. These are obtained by a perturbation argument starting from a simpler model. Unfortunately we could not decide analytically whether the periodic solutions are stable or unstable. Simulations indicate that they are stable at least in some cases. , where
, where 